“Cellular senescence and disrupted proteostasis induced by myotube atrophy are prevented with low-dose metformin and leucine cocktail.”
BUFFALO, NY- March 31, 2023 – A new research paper was published on the cover of Aging (listed by MEDLINE/PubMed as “Aging (Albany NY)” and “Aging-US” by Web of Science) Volume 15, Issue 6, entitled, “Cellular senescence and disrupted proteostasis induced by myotube atrophy are prevented with low-dose metformin and leucine cocktail.”
Aging coincides with the accumulation of senescent cells within skeletal muscle that produce inflammatory products, known as the senescence-associated secretory phenotype, but the relationship of senescent cells to muscle atrophy is unclear. Previously, researchers found that a metformin + leucine (MET+LEU) treatment had synergistic effects in aged mice to improve skeletal muscle structure and function during disuse atrophy.
In this new study, researchers Jonathan J. Petrocelli, Naomi M.M.P. de Hart, Marisa J. Lang, Elena M. Yee, Patrick J. Ferrara, Dennis K. Fix, Amandine Chaix, Katsuhiko Funai, and Micah J. Drummond from the University of Utah aimed to determine the mechanisms by which MET+LEU exhibits muscle atrophy protection in vitro and if this occurs through cellular senescence.
“The purpose of this study was to identify the skeletal muscle cell-intrinsic effects of MET+LEU during an atrophy stimulus. Secondarily, we sought to determine the possible mechanisms underlying MET+LEU action on skeletal muscle cells with an emphasis on cellular senescence.”
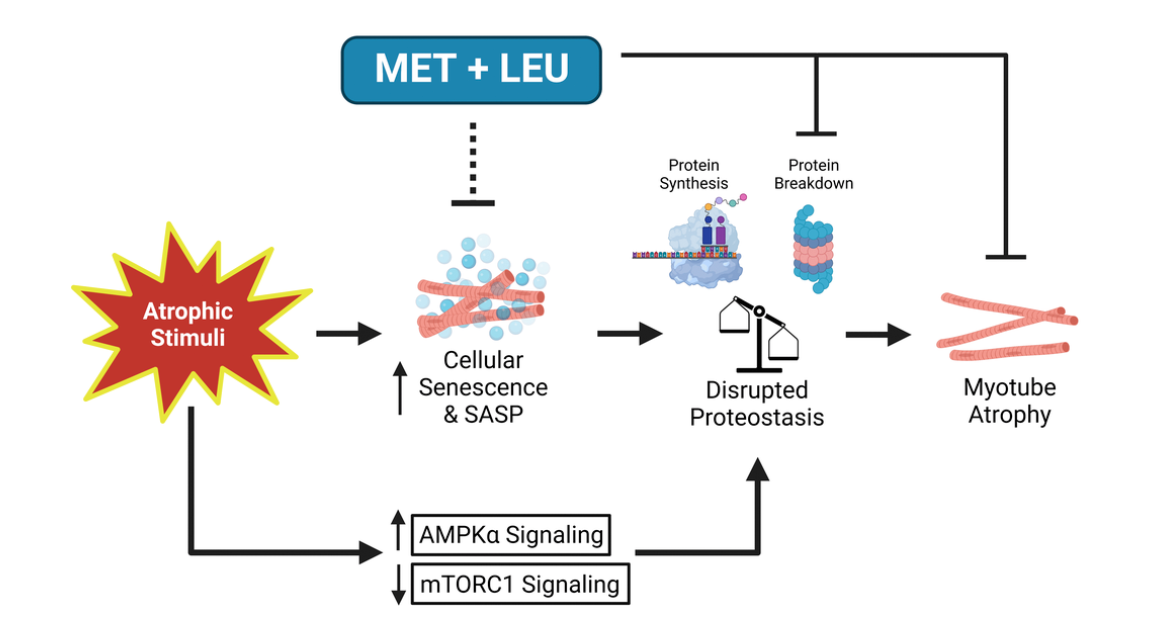
C2C12 myoblasts differentiated into myotubes were used to determine MET+LEU mechanisms during atrophy. Additionally, aged mouse single myofibers and older human donor primary myoblasts were individually isolated to determine the translational potential of MET+LEU on muscle cells. MET+LEU (25 + 125 μM) treatment increased myotube differentiation and prevented myotube atrophy. Low concentration (0.1 + 0.5 μM) MET+LEU had unique effects to prevent muscle atrophy and increase transcripts related to protein synthesis and decrease transcripts related to protein breakdown. Myotube atrophy resulted in dysregulated proteostasis that was reversed with MET+LEU and individually with proteasome inhibition (MG-132).
Inflammatory and cellular senescence transcriptional pathways and respective transcripts were increased following myotube atrophy yet reversed with MET+LEU treatment. Dasatinib + quercetin (D+Q) senolytic prevented myotube atrophy similar to MET+LEU. Finally, MET+LEU prevented loss in myotube size in alternate in vitro models of muscle atrophy as well as in aged myofibers while, in human primary myotubes, MET+LEU prevented reductions in myonuclei fusion. These data support that MET+LEU has skeletal muscle cell-autonomous properties to prevent atrophy by reversing senescence and improving proteostasis.
“In conclusion, this study provides evidence of a possible link between cellular senescence and disrupted proteostasis that is targeted by MET+LEU in muscle cells to reverse the muscle atrophy phenotype.”
DOI: https://doi.org/10.18632/aging.204600
Corresponding Author: Micah J. Drummond – micah.drummond@hsc.utah.edu
Keywords: skeletal muscle atrophy, inflammation, senolytic, AMPK, protein breakdown
Sign up for free Altmetric alerts about this article: https://aging.altmetric.com/details/email_updates?id=10.18632%2Faging.204600
About Aging-US:
Launched in 2009, Aging (Aging-US) publishes papers of general interest and biological significance in all fields of aging research and age-related diseases, including cancer—and now, with a special focus on COVID-19 vulnerability as an age-dependent syndrome. Topics in Aging go beyond traditional gerontology, including, but not limited to, cellular and molecular biology, human age-related diseases, pathology in model organisms, signal transduction pathways (e.g., p53, sirtuins, and PI-3K/AKT/mTOR, among others), and approaches to modulating these signaling pathways.
Please visit our website at www.Aging-US.com and connect with us:
Click here to subscribe to Aging publication updates.
For media inquiries, please contact media@impactjournals.com.

{kind=link}